In this post I’ll cover how to run f4-statistics using the admixtools package for R. While I do not typically use R for general-purpose programming, I prefer this implementation over the original one because working in a REPL environment is more practical than editing parameter files, especially when you’re testing different population combinations. The interactive workflow makes programmatic model testing straightforward. Beyond these workflow improvements, the R version is also significantly faster, not because of the language itself, but simply better implementation. You can work with the full AADR dataset without creating subsets.
Setting Up R and Admixtools
First, install R and the required dependencies. For Ubuntu/Debian:
sudo apt update -y
sudo apt install -y r-base r-base-dev build-essential \
libcurl4-openssl-dev libssl-dev libxml2-dev libgsl-dev
Next, start R by running R in your terminal, then install admixtools:
install.packages("remotes")
remotes::install_github("uqrmaie1/admixtools")
What are f4-statistics?
F4-statistics measure genetic relationships between four populations by computing the average of (a-b)*(c-d) across SNPs, where a, b, c, d are allele frequencies in populations A, B, C, D respectively. They test whether allele frequency patterns are consistent with a simple tree topology, or whether they show evidence of admixture or differential relatedness between specific population pairs.
The statistic is written as F4(A, B; C, D), and under a simple tree model with no admixture, this value should be zero. In practice, the observed value is never exactly zero due to variation, which is why we use z-scores to assess statistical significance rather than expecting literal zeros.
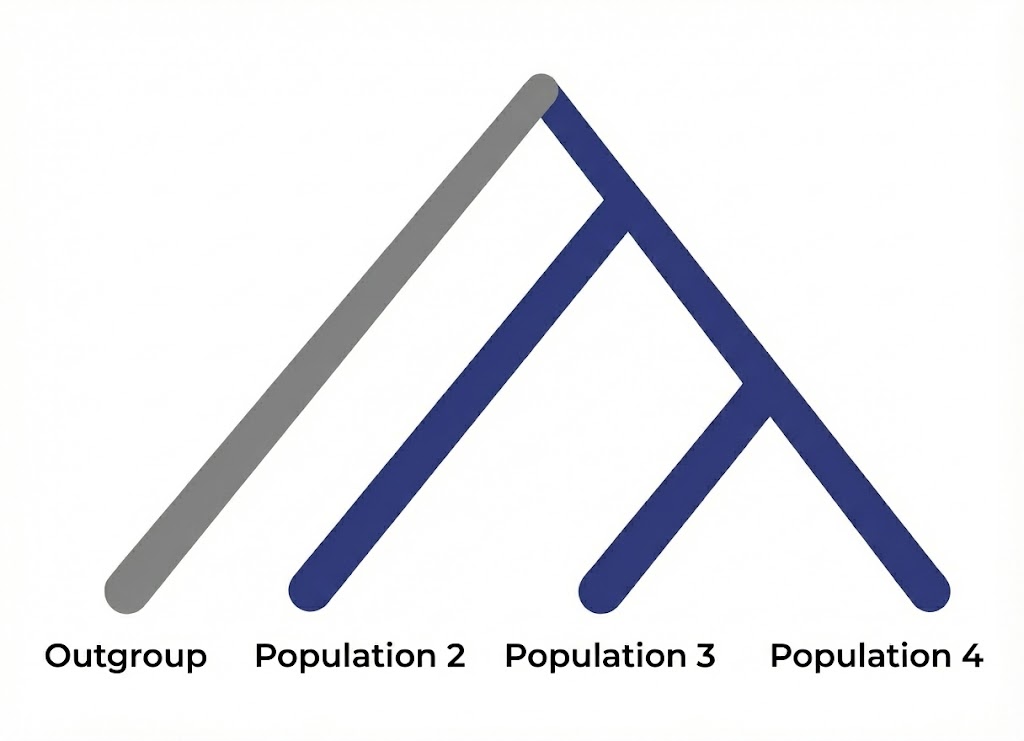
In the assumed tree topology, this is because any drift that affects the (A, B) branch should be independent of drift on the (C, D) branch. When F4 significantly deviates from zero, it indicates that the four populations don’t fit a simple tree, usually because of gene flow between populations that shouldn’t be connected under the assumed topology.
This might sound more complicated than it is. In essence you’re testing whether populations form a simple tree or show deviations consistent with gene flow between populations.
Interpreting f4-statistics (with A as deep outgroup): When using a deep outgroup in position A (like Mbuti), the sign tells you which populations share more drift:
- Positive f4(A,B;C,D): Population B shares more drift with D than with C
- Negative f4(A,B;C,D): Population B shares more drift with C than with D
- Zero (or non-significant): B shares equal drift with C and D, consistent with a simple tree
Statistical significance is assessed using z-scores. The conventional threshold is |z| > 3, though |z| > 2 could also be used.
Running f4-statistics
I’ll use the AADR dataset, the most comprehensive curated collection of ancient and modern genomic samples. Since Admixtools works with EIGENSTRAT files and AADR is distributed in this format, the dataset can be used directly without preprocessing. For download instructions, see: Download Ancient & Modern DNA (AADR Tutorial).
Next, navigate to the directory containing your downloaded AADR dataset files (.geno, .snp, .ind) and start R:
# move into the folder so the files can be loaded without specifying paths
cd /path/to/aadr/dataset
R
Load the previously installed admixtools library and dataset:
library(admixtools)
# Specify the file prefix (without extensions)
# If your files are named v62.0_HO_public.geno/.snp/.ind
# use only the prefix:
data = "v62.0_HO_public"
When specifying populations in f4-statistics, use the exact population labels from the third column of your .ind file, the function will automatically include all samples assigned to that population label.
Now we can run f4-statistics. You’ll need specific hypotheses about population relationships to test. I’ll demonstrate three examples that illustrate different outcomes.
In the examples below, pop1 through pop4 correspond to populations A, B, C, and D respectively, where (A, B) and (C, D) form the two pairs being compared.
Example 1: Do Yamnaya Share More Drift with EHG than WHG?
For this test, we’ll use Mbuti as the outgroup (position A), Yamnaya in position B, and EHG and WHG in positions C and D respectively. Note that positions within pairs (A,B) and (C,D) are interchangeable. Swapping the members of either pair reverses the sign of the f4 value, since the test then measures the opposite direction of excess allele sharing. If you are unsure about orientation, just follow the arrangement used in the examples below, where the sample of interest is placed in position B.
f4(data, pop1="Mbuti.HO", pop2="Russia_Samara_EBA_Yamnaya.AG", pop3="Italy_Epigravettian_alt.AG.BY.AA", pop4="Latvia_Mesolithic_oEHG.AG")
The result shows:
pop1 pop2 pop3 pop4 est se z p n
<chr> <chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Mbuti.HO Russia_Samara_EBA_Ya… Ital… Latv… 0.00245 9.17e-4 2.67 0.00751 49477
The z-score of 2.67 falls just below the conventional significance threshold of 3, but the result is still informative. While some use a stricter threshold of |z| > 3, others consider |z| > 2 sufficient for exploratory analysis, particularly when the direction of the effect is theoretically motivated. The positive value indicates that Yamnaya shares more drift with EHG than with the WHG sample from Villabruna.
I should note that this test was run on an AADR SNP subset. Using the full AADR-HO or 1240k dataset would likely increase the signal and push |z| above 3. Likewise, substituting a more standard EHG reference such as Samara HG would probably produce a clearly significant result. For demonstration purposes, however, this borderline case is useful because it shows how to interpret near-threshold values.
The output also reports n, which is the number of SNPs used in the calculation. Here, roughly 49k SNPs contributed to the statistic. In general, larger SNP counts increase accuracy, making it easier to reach |z| > 3.
Example 2: Do Anatolian Neolithic Farmers Share More Drift with Sardinians or French?
f4(data, pop1="Mbuti.HO", pop2="Turkey_Marmara_Barcin_N.AG", pop3="Sardinian.HO", pop4="French.HO")
The result shows:
pop1 pop2 pop3 pop4 est se z p n
<chr> <chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Mbuti.HO Turkey_Marmara_Ba… Sard… Fren… -0.00233 1.25e-4 -18.6 2.49e-77 155527
This result is highly significant with a z-score of -18.6. The negative value indicates that Anatolian Neolithic farmers (population B) share significantly more drift with Sardinians (population C) than with the French (population D). This is consistent with Sardinians retaining the highest proportion of Neolithic farmer ancestry in Europe
It’s important to note that the interpretation of an f4-statistic depends entirely on the populations you choose. If we replaced Sardinians with Han Chinese, the result would again be highly significant and show strong drift sharing between Neolithic Farmers and the French, but this time it would reflect shared Western Eurasian ancestry at a much deeper time depth, rather than specifically Neolithic farmer ancestry.
Example 3: How Do French, Scottish, and Norwegians Relate to Each Other?
f4(data, pop1="Mbuti.HO", pop2="French.HO", pop3="Scottish.HO", pop4="Norwegian.HO")
The result shows:
pop1 pop2 pop3 pop4 est se z p n
<chr> <chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Mbuti.DG French.HO Scottish.HO Norwegian.HO 1.92e-4 2.05e-4 0.941 0.347 155583
The f4-statistic is non-significant (z = 0.941), indicating that the French share equal amounts of drift with Scottish and Norwegian populations. This result is consistent with a simple tree topology where these three populations diverged without subsequent gene flow between specific pairs. In other words, their genetic relationships can be explained by shared ancestry and geographic distance, without requiring admixture events that would preferentially connect any two of them.
Conclusion
F4-statistics are excellent for detecting signals of admixture and differential relatedness, but be cautious about overinterpreting the results. A significant f4-statistic tells you that populations don’t fit a simple tree and indicates excess allele sharing between specific pairs, but it doesn’t tell you the direction of gene flow, whether the relationship is direct or through an intermediate population (ancestral to both), or the ancestry proportions involved. For that, you need qpAdm, which I’ll cover in the next post.